




Farmacovigilanza
La normativa europea in materia di Farmacovigilanza è stata modificata con l’adozione nel 2010, del Regolamento UE 1235/2010, la cui applicazione è operativa dal 2 luglio 2012, e della Direttiva 2010/84/UE, attualmente in fase di recepimento.
In primo luogo, cambia la definizione di reazione avversa (ADR) intesa ora come “Effetto nocivo e non voluto conseguente all’uso di un medicinale”. Di fatto, con tale definizione, che è indipendente dal tipo di uso del medicinale, devono essere oggetto di segnalazione tutte le reazioni avverse, incluse quelle derivanti da errore terapeutico, abuso, inefficacia, uso off label, sovradosaggio ed esposizione professionale. L'obiettivo è un incremento delle segnalazioni a cui dovrà corrispondere una maggiore attività di monitoraggio
E’ stato stimato che il 5% di tutti gli accessi in ospedale sono dovuti a reazioni avverse (ADRs), che il 5% di tutti i pazienti già ricoverati in ospedale presenta una ADR, che le ADRs sono al quinto posto tra le cause di morte in ospedale. Pertanto, si è reso necessario intervenire sulle normative in vigore al fine di promuovere e proteggere la salute pubblica riducendo il numero e la gravità delle ADRs e migliorando l’uso dei medicinali attraverso diversi tipi di intervento.
Fondamentalmente, i cambiamenti introdotti tendono ad aumentare l’efficacia, la rapidità e la trasparenza degli interventi di Farmacovigilanza attraverso regole che mirano a:
· rafforzare i sistemi di Farmacovigilanza, (ruoli e responsabilità chiaramente definiti per tutte le parti )
· razionalizzare le attività tra gli Stati Membri ad esempio attraverso una ripartizione delle stesse attività con condivisione del lavoro svolto evitando duplicazioni ,
· incrementare la partecipazione dei pazienti e degli operatori sanitari,
· migliorare i sistemi di comunicazione delle decisioni prese e darne adeguata motivazione,
· aumentare la trasparenza.
In tutti i paesi dell’UE i pazienti possono segnalare direttamente le sospette reazioni avverse. In Italia questa possibilità è già prevista da anni mediante modulo cartaceo, ma oggi, in accordo anche alla nuova direttiva, le segnalazioni possono essere effettuate anche via web.
Tutte le segnalazioni di reazioni avverse confluiscono nel database europeo Eudravigilance ma con una tempistica diversificata a seconda della gravità della reazione (entro 15 giorni per le segnalazioni gravi ed entro 90 giorni per quelle non gravi) .I dati delle reazioni avverse saranno resi accessibili e per alcuni medicinali autorizzati all’immissione in commercio con procedura centralizzata europea è già possibile consultare il database europeo delle reazioni avverse collegandosi all’indirizzo www.adrreports.eu.
La nuova normativa è improntata anche ad una maggiore trasparenza e a migliorare la comunicazione. Infatti sono resi disponibili al pubblico, attraverso il portale web dell'AIFA:
1. rapporti di valutazione pubblici, unitamente a una loro sintesi;
2. riassunti delle caratteristiche del prodotto e fogli illustrativi;
3. riassunti dei piani di gestione del rischio;
4. elenco dei medicinali sottoposti a monitoraggio addizionale;
5. informazioni sulle diverse modalità per la segnalazione di sospette reazioni avverse dei medicinali alle autorità competenti da parte degli operatori sanitari e dei pazienti, compresi i moduli con maschera web di inserimento dati.
I medicinali sottoposti a monitoraggio addizionale sono i prodotti contenenti nuove sostanze attive non presenti in medicinali autorizzati in Europa alla data del 1 gennaio 2011; tra questi biologici e biosimilari, ma possono essere inclusi anche i prodotti la cui autorizzazione è subordinata a particolari condizioni o autorizzati in circostanze eccezionali; i prodotti soggetti a studi sulla sicurezza dopo la concessione dell'autorizzazione all'immissione in commercio AIC.
Questi medicinali sottoposti a monitoraggio addizionale sono identificabili dal foglio illustrativo che reca la dicitura “Medicinale sottoposto a monitoraggio addizionale” preceduta da un simbolo nero, il cui elenco è stilato dall’Agenzia Europea dei Medicinali.
Il sistema previsto dalla nuova legislazione ed il suo funzionamento sono piuttosto complessi ed è necessaria una congrua dotazione di personale competente adeguatamente qualificato e addestrato, come specificato anche nel Regolamento di Esecuzione (UE) 520/2012 del 19 giugno 2012.
Le premesse per il raggiungimento di tali obiettivi sono state già poste in essere, mentre, per quanto riguarda i risultati bisognerà attendere il pieno funzionamento di quanto previsto, anche in termini di formazione del personale sanitario, con particolare riguardo alle attività di Farmacovigilanza da parte delle strutture sanitarie e, più in generale, una maggiore partecipazione di tutte le parti interessate, inclusi i pazienti.
La Farmacovigilanza è l'insieme delle attività il cui obiettivo è quello di fornire, in modo continuativo, le migliori informazioni possibili sulla sicurezza dei farmaci permettendo così l'adozione delle misure opportune e in tal modo assicurare che i farmaci disponibili sul mercato presentino, nelle condizioni di utilizzo autorizzate, un rapporto beneficio rischio favorevole per la popolazione (v. D.Lvo 95/03 "Attuazione della direttiva 2000/38/CE relativa alle specialità medicinali).
La farmacovigilanza ha quattro obiettivi principali:
1. riconoscere, il più rapidamente possibile, nuove reazioni avverse ai farmaci
2. migliorare ed allargare le informazioni su reazioni sospette o già note
3. valutare i vantaggi di un farmaco su altri o su altri tipi di terapia
4. comunicare l'informazione in modo da migliorare la pratica terapeutica
Le nuove disposizioni emanate a livello Europeo (regolamento UE 1235/2010 e direttiva 2010/84/UE) ed implementate a luglio del 2012, hanno ridefinito gli obiettivi in materia di farmacovigilanza per promuovere e proteggere la salute pubblica riducendo il numero e la gravità delle reazioni avverse e migliorando l’uso dei medicinali tramite:
- Ruoli e responsabilità di tutte le parti coinvolte
- Assicurazione di un sistema europeo robusto e rapido nel prendere le decisioni necessarie in materia di FV
- Incremento della partecipazione dei pazienti e degli operatori sanitari
- Miglioramento dei sistemi di comunicazione sulle decisioni prese e loro giustificazione mediante:
- Aumento di trasparenza
- Migliore informazione sui medicinali
- Aumento dell’efficienza dei sistemi di FV
- Rafforzamento della Rete Europea di FV
- Rafforzamento dei Sistemi di FV aziendali
- Aumentata proattività/programmazione delle attività da condurre
La farmacovigilanza comprende una serie di attività che coinvolgono le Istituzioni (EMA, AIFA), i produttori di farmaci, le strutture e il personale sanitario (es. medici, farmacisti, infermieri), i pazienti. Anche il contributo del cittadino è essenziale, ogni cittadino dovrebbe infatti essere sensibilizzato il più possibile a segnalare al proprio medico o al farmacista l'eventuale comparsa di qualsiasi effetto collaterale, anche di quelli indicati nel foglietto illustrativo, poiché ciò permette di compilare statistiche precise sul medicinale. Quanto meglio ciascuno di questi attori svolge la propria funzione, tanto più è garantita la sicurezza dei farmaci in commercio.
Tutti i componenti sono connessi fra loro in una rete, la Rete Nazionale di Farmacovigilanza, che è gestita dall’AIFA e ad oggi comprende tutte le Regioni italiane, tale sistema nazionale è utilizzato per raccogliere informazioni sui rischi dei medicinali in relazione alla salute dei pazienti o alla salute pubblica.
Le informazioni si riferiscono in particolare alle reazioni avverse nell’uomo, derivanti sia dall’utilizzo del medicinale conformemente alle condizioni contenute nell’AIC sia dall’uso al di fuori delle condizioni di autorizzazione in questione (OFF-LABEL), nonché alle reazioni avverse associate all’esposizione per motivi professionali.
Le regioni, singolarmente o di intesa fra loro, collaborano con l'AIFA nell'attività di farmacovigilanza, fornendo elementi di conoscenza e valutazione ad integrazione dei dati oltre a fornire i dati sui consumi dei medicinali mediante programmi di monitoraggio sulle prescrizioni a livello regionale, e provvedono, nell'ambito delle proprie competenze, alla diffusione delle informazioni al personale sanitario ed alla formazione degli operatori nel campo della farmacovigilanza.
Le strutture sanitarie come: aziende sanitarie locali, aziende ospedaliere, istituti di ricovero e cura a carattere scientifico pubblici e privati, policlinici universitari pubblici e privati e altre analoghe strutture, nominano una persona adeguatamente qualificata responsabile della farmacovigilanza, che provvede a registrarsi alla rete nazionale di farmacovigilanza al fine dell'abilitazione necessaria per la gestione delle segnalazioni.
Le strutture sanitarie private, al fine di assolvere ai compiti di farmacovigilanza, fanno riferimento alla persona qualificata responsabile della farmacovigilanza della azienda sanitaria locale competente per territorio.
PRINCIPALI CRITICITA’ NEL SISTEMA DI FARMACOVIGILANZA
L'efficienza dei sistemi di segnalazione spontanea di ADR si fonda sui presupposti che:
- un evento avverso che si verifica in un paziente in trattamento con uno o più farmaci possa essere riconosciuto come tale;
- che possa essere sospettata l'esistenza di un rapporto causale tra insorgenza dell'evento e assunzione di un farmaco;
- che l'evento venga segnalato.
A quest’ultimo proposito William Inman formulò i sette peccati mortali dei medici, vale a dire le cause per le quali i medici non segnalano le ADR:
Complacency
L'erronea convinzione che vengono commercializzati soltanto farmaci 'sicuri'
Fear
Timore di essere coinvolti in cause legali
Guilt
Senso di colpa per aver causato danni al paziente a causa del trattamento prescritto
Ambition
Desiderio di raccogliere e pubblicare una casistica personale
Ignorance
Ignoranza delle procedure per la segnalazione
Diffidence
Timore di segnalare sulla base di sospetti che potrebbero rivelarsi infondati
Lethargy
Un insieme di tendenza a procrastinare la segnalazione, disinteresse, mancanza di tempo, indisponibilità del modulo di segnalazione, ecc.
COSA E’ UNA REAZIONE AVVERSA?
Nel corso degli ultimi decenni sono state proposte varie definizioni per descrivere una Reazione Avversa da Farmaco (ADR, dall’inglese Adverse Drug Reaction), tuttavia nessuna di queste, benché formalmente corretta, è riuscita ad esprimere in maniera esaustiva il concetto a causa delle criticità che emergono nella interpretazione della definizione stessa.
Ad esempio, la definizione di Laurence che escludeva in maniera specifica le reazioni indesiderate di lieve entità (es. una leggera secchezza delle fauci), definiva una reazione avversa come:
“Un effetto dannoso o sgradevole causato da un farmaco alle dosi terapeutiche, che giustifica la riduzione della dose o la sospensione del farmaco stesso e/o ne predice il rischio derivante da una futura somministrazione”; tuttavia questa definizione non contempla l’errore come causa di un effetto avverso, cionondimeno esclude la possibilità che la reazione sia dovuta a contaminanti (es., nei prodotti erboristici) o ad eccipienti “presumibilmente” inattivi presenti all’interno della formulazione.
Allo stato attuale, a seguito della nuova normativa Europea in materia di farmacovigilanza (regolamento UE 1235/2010 e direttiva 2010/84/UE) la reazione avversa è stata ridefinita come:
“Effetto nocivo e non voluto conseguente all’uso di un medicinale conformemente alle indicazioni contenute nell’autorizzazione all’immissione in commercio, agli errori terapeutici, agli usi non conformi alle indicazioni contenute nell’autorizzazione all’immissione in commercio, incluso il sovradosaggio, l’uso improprio, l’abuso del medicinale, nonché associato all’esposizione per motivi professionali ”.
Inoltre, è bene puntualizzare alcuni aspetti riguardanti le differenze tra termini che in questo ambito vengono spesso “erroneamente” utilizzati come sinonimi:
· “Reazione Avversa” ed “Effetto avverso” sono di fatto intercambiabili, entrambi sono riferibili ad un esito dannoso da attribuire all’azione del farmaco ad eccezione del fatto che l’effetto avverso è considerato dal punto di vista del farmaco, mentre la reazione avversa è considerata dal punto di vista del paziente;
· “Evento Avverso” è un esito indesiderato che si verifica mentre un paziente è in fase di trattamento con un farmaco, ma non è necessariamente correlato all’assunzione del farmaco stesso;
· “Effetto Tossico” è da riferirsi ad un’esagerazione del normale effetto farmacologico e non è comune alle normali dosi terapeutiche (es. una cefalea causata dall’utilizzo di un calcio-antagonista derivante dall’esasperazione del normale meccanismo d’azione di vasodilatazione del farmaco stesso), infatti un effetto tossico è sempre dose-dipendente;
· “Effetto collaterale” è un esito che si verifica attraverso qualche meccanismo d’azione del farmaco (non necessariamente quello per cui il farmaco viene somministrato) e può essere o no dose-dipendente, peraltro è importante sottolineare che tale effetto potrebbe essere non desiderabile ma, talora può risultare anche favorevole (es. l’utilizzo di un beta-bloccante per il trattamento dell’ipertensione può dare sollievo dal dolore anginoso);
· “Reazione avversa inattesa” è una reazione la cui natura e severità non è riportata nel foglietto illustrativo o nella autorizzazione alla commercializzazione del farmaco o che sia inattesa rispetto alle caratteristiche del farmaco stesso;
· “Reazione avversa inattesa o evento avverso serio” un evento medico spiacevole che, per qualsiasi dose: metta in pericolo la vita del paziente, richieda l'ospedalizzazione del paziente o prolunghi una ospedalizzazione già avvenuta; determini una persistente o significativa disabilità o incapacità; provochi la morte; comporta una anomalia congenita o un difetto alla nascita.
CLASSIFICAZIONE DELLE REAZIONI AVVERSE
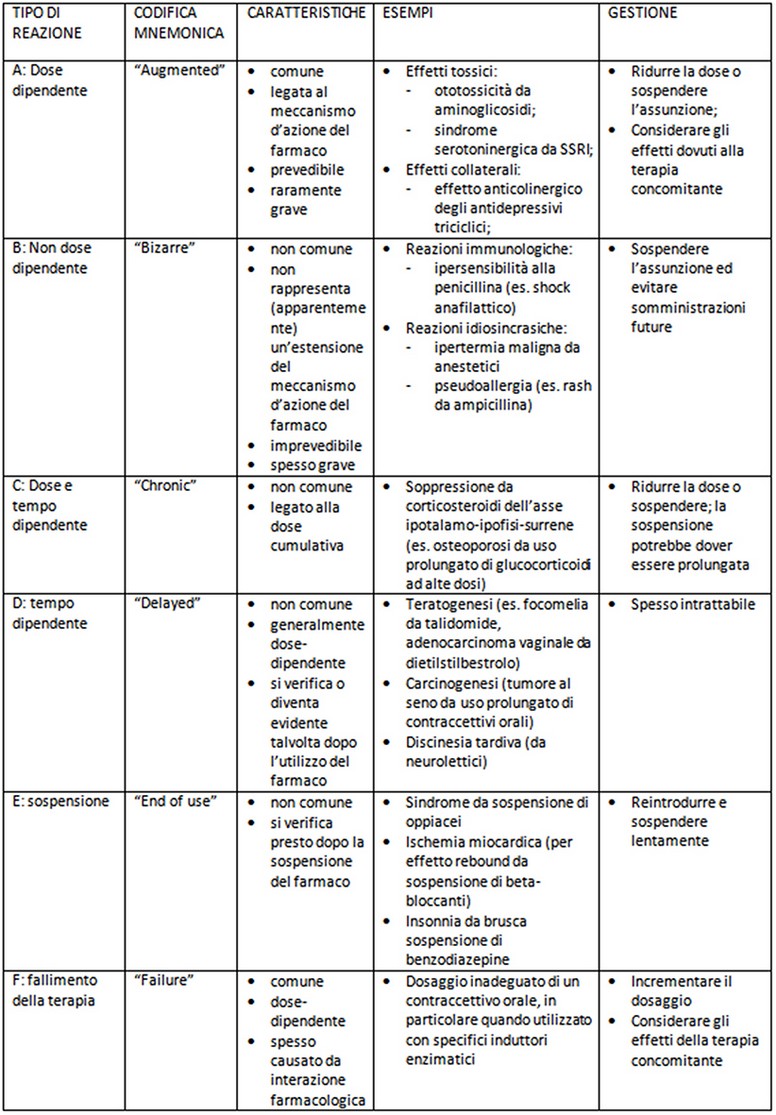
VALUTAZIONE DEL NESSO DI CAUSALITA’
fra farmaco e reazione avversa è classificata dal WHO International Drug Monitoring Programme come:
¨ CERTA
Quando è un evento (inclusa l'alterazione di un parametro di laboratorio) che insorge con una plausibile sequenza temporale dall'assunzione del farmaco e che non può essere spiegato dalla malattia per il quale il farmaco è impiegato, né dalla concomitante assunzione di altri farmaci o sostanze chimiche.
La reazione deve migliorare con il dechallange e ricomparire con il rechallange.
¨ PROBABILE
Quando è un evento (inclusa l'alterazione di un parametro di laboratorio) che insorge con una plausibile sequenza temporale dall'assunzione del farmaco e che non può essere spiegato dalla malattia per il quale il farmaco è impiegato, né dalla concomitante assunzione di altri farmaci o sostanze chimiche.
La reazione deve migliorare con il dechallange.
Non è necessario il rechallange perché la reazione avversa rientri in questa definizione.
¨ POSSIBILE
Quando è un evento (inclusa l'alterazione di un parametro di laboratorio) che insorge con una plausibile sequenza temporale dall'assunzione del farmaco e che non può essere spiegato dalla malattia per il quale il farmaco è impiegato, né dalla concomitante assunzione di altri farmaci o sostanze chimiche.
Non è necessario avere informazioni sull'effetto del dechallange.
¨ IMPROBABILE
Quando è un evento (inclusa l'alterazione di un parametro di laboratorio) che insorge con una sequenza temporale dall'assunzione del farmaco che rende improbabile una connessione causale o in cui altri farmaci o sostanze chimiche o la malattia del paziente possono spiegare l'evento osservato.
¨ CONDIZIONATA/INCLASSIFICATA
Quando è un evento (inclusa l'alterazione di un parametro di laboratorio) riportato come una reazione avversa, che necessita di più dati per un’appropriata valutazione o per la quale si stanno valutando ulteriori dati.
¨ NON VALUTABILE/INCLASSIFICABILE
Una segnalazione che riporta una reazione avversa che non può essere giudicata a causa della insufficienza o contraddittorietà delle informazioni e che non può essere verificata o supportata da altre informazioni.
Normativa nazionale:
· Decreto Ministero della Salute 30 aprile 2015 - Procedure operative e soluzioni tecniche per un’efficace azione di Farmacovigilanza adottate ai sensi del comma 344 dell’articolo 1 della legge 24 dicembre 2012, n. 228 (Legge di stabilità 2013).
· Decreto Legislativo 24 aprile 2006, n. 219 e s.m.i. - Attuazione della direttiva 2001/83/CE (e successive direttive di modifica) relativa ad un codice comunitario concernente i medicinali per uso umano, nonché della direttiva 2003/94/CE.
· Decreto Legislativo 29 dicembre 2007 (Disposizioni correttive al decreto legislativo 24 aprile 2006, n. 219, recante attuazione della direttiva 2001/83/CE relativa ad un codice comunitario concernente medicinali per uso umano).
· Legge 24 dicembre 2012, n. 228 - Disposizioni per la formazione del bilancio annuale e pluriennale dello Stato (Legge di stabilità 2013)
· Decreto Legislativo 4 marzo 2014, n. 42 - Attuazione dell'articolo 1, paragrafi 1, 5 e 12 della direttiva 2012/26/UE, che modifica la direttiva 2001/83/CE, per quanto riguarda la Farmacovigilanza.
Normativa comunitaria: